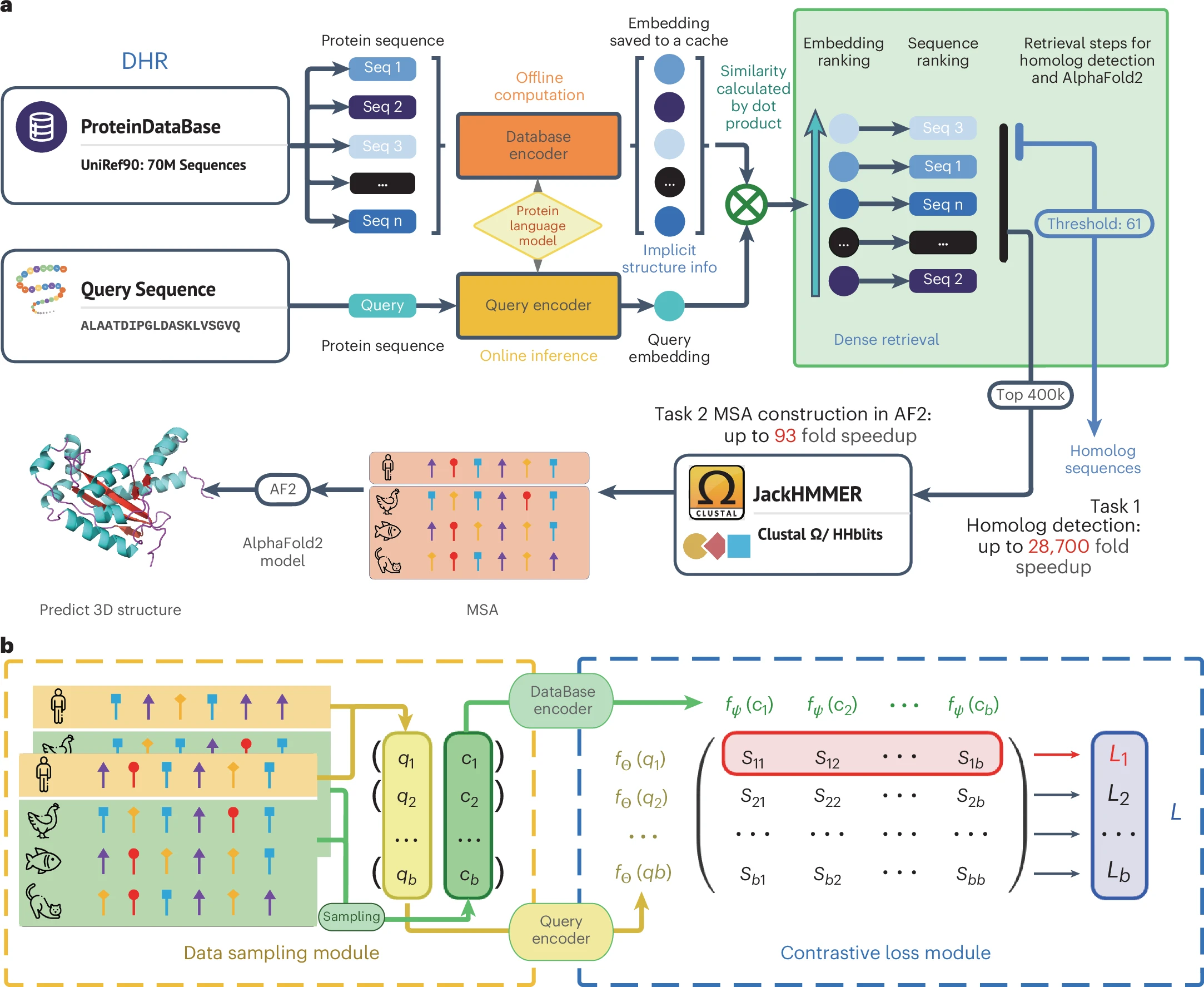
DHR – a rapid pipeline for alignment-free protein homolog detection
A research team led by Professor Yu Li from the Department of Computer Science and Engineering at the Chinese University of Hong Kong (CUHK) has developed a ground-breaking method, Dense Homolog Retrieval (DHR), for the rapid and sensitive detection of remote protein homologs from extensive databases. The study was published in Nature Biotechnology in August 2024. The research was led by two PhD students Liang Hong and Zhihang Hu in Prof. Yu Li’s group. Zhihang Hu is also co-supervised by Prof. Irwin King. This work started from Auguet 2021 when Liang was a Year-3 UG student, and, initially, it was Liang’s UG Final Year Project done in Professor Yu Li’s group .
Revolutionizing Protein Homolog Detection
Protein homolog detection is a crucial step in computational biology, essential for numerous biological sequence-related research areas, including protein structure prediction, biomolecular functional analysis, transcription regulation studies, and phylogenetic reconstruction. With the advent of next-generation sequencing technologies, the volume of biological sequences has surged, creating a demand for ultra-fast and highly sensitive methods to enhance our understanding of protein evolution, structure, and function.
Dense Homolog Retrieval
DHR leverages the power of protein language models and dense retrieval techniques to offer unmatched efficiency in homolog detection. This novel pipeline exhibits a consistent increase in sensitivity across various benchmarks while being significantly faster than traditional methods. When detecting protein homologs, it achieves a more than 10% increase in sensitivity compared with previous methods and an over 56% increase in sensitivity at the superfamily level for samples that are challenging to identify using alignment-based approaches. DHR is up to 22 times faster than traditional methods such as PSI-BLAST and DIAMOND2 and up to 28,700 times faster than HMMER.
Performance Highlights
- Sensitivity and Speed: DHR not only matches but surpasses traditional methods in sensitivity, achieving superior results in detecting diverse and remote homologs. It achieves a more than 10% increase in sensitivity compared with previous methods and an over 56% increase in sensitivity at the superfamily level for samples that are challenging to identify using alignment-based approaches.
- Efficiency: The method is tens of times faster, enabling rapid analysis and processing of large-scale sequence data. It is up to 22 times faster than traditional methods such as PSI-BLAST and DIAMOND2 and up to 28,700 times faster than HMMER.
- MSA Generation: When combined with sequence alignment modules, DHR can instantly generate MSAs almost identical to those produced by AlphaFold2’s default pipeline. Moreover, when integrated with JackHMMER, DHR enhances MSA diversity and comprehensiveness, thereby improving structure prediction accuracy and supporting various downstream applications. For example, it can promote the protein prediction accuracy by 0.4 Å root mean square deviation (a measure of similarity between two superimposed atomic coordinates, commonly used to compare protein structures) on average when merged with AlphaFold2 default MSAs.
Future Prospects
The development of DHR represents a significant leap forward in computational biology, offering a powerful tool for large-scale protein-related research. As the field continues to evolve, the adoption of such advanced methods will be crucial in uncovering new insights into protein function and evolution.
About Nature Biotechnology
Nature Biotechnology originates from 1983 and is the most prestigious journal across the biology and computational biology field, with an Impact Factor (IF) of 33.1 and 5-year IF=56.9.
For more details, DHR is published in Nature Biotechnology